6.2 Efektor HPV
6.2.1 Umístění efektoru
Hlavním smyslem HPV je zabezpečení optimálního okysličení krve prostřednictvím udržování potřebného poměru ventilace/perfúze. Proto lze při hypoxii očekávat největší vazokonstrikci spíše
na arteriální straně plicního řečiště. Pokud by totiž místem nejsilnější
HPV byly plicní žíly, vedla by hypoxie ke zvýšení kapilárního tlaku
a v důsledku toho i kapilární filtrace. Vzniklý edém by mohl narušit
difúzi plynů a okysličování krve by se naopak zhoršilo. Pokusně
bylo zjištěno, že při akutní hypoxii skutečně nedochází ke zvýšenému
prosakování tekutiny z plicních kapilár [1, 2, 3]. Hypoxie tedy asi nepůsobí příliš silnou plicní venokonstrikci.
Řada údaju ukazuje, že cévy, které se při akutní hypoxii kontrahují,
jsou hlavně na arteriální straně plicního řečiště. Objem krve
v nich klesá [4, 5, 6]. Objem krve v kapilárách a žilách lehce stoupá [4]. Při retrográdní perfúzi plic vede HPV, na rozdíl od plic perfundovaných ortográdně, k progresivní akumulaci
vody [7, 8, 9].
Katetrizační měření u zvířat [10, 11, 12, 13] i lidí [14, 15, 16] ukazují, že se při akutní hypoxii zvyšuje tlak v plícnici, ale
tlak v katetru zaklíněném v malé segmentární plicní tepně se podstatně
nemění. Tlak v zaklínění odpovídá tlaku v plicních žilách [17, 18]. Ani tlak měřený katetrem přímo v malé plicní žíle a v levé
síni se při hypoxii nemění [19, 20]. Při hypoxii tedy roste odpor proximálně od malých žil.
Přímá měření změn průsvitu různých plicních cév a tlaku v nich
i měření in vitro ukazují, že se při akutní hypoxii kontrahují především malé arterioly.
Hirschman a Bouncek [21] pozorovali při plicní angiografii u psů zmenšení průsvitu malých
artérií za hypoxie, zatímco větší proximálnejší se poněkud rozšiřovaly.
Podobné pozorování publikovali Allison a Stanbrook [22]. Kato a Staub [23] rychle zmrazili plíce při HPV vyvolané v jednom laloku. Histologické vyšetření pak ukázalo
zúžení vnitřního průměru arteriol terminální respirační jednotky.
Při vystavení části povrchu žabích plic dusíku bylo pomocí laserové
mikroskopie in vivo pozorováno zmenšení průsvitu arteriol hypoxické
oblasti a zpomalení průtoku krve [24]. Shirai se spolupracovníky ukázali na kočkách pomocí rentgenového
televizního systému zmenšení průsvitu arteriol o průměru 100-600
um závislé na stupni hypoxie a doprovázené snížením rychlosti
proudu a objemového průtoku [25]. Podobně i u fretek se při hypoxii zužují cévy s průměrem menším
než cca 600 um (zatímco větší cévy se spíše rozšiřují, pravděpodobně
v důsledku kombinace slabší vlastní konstrikce a zvýšeného odporu
v mikrocirkulaci) [89]. Laboratoř Dr. Wagnera nedávno pomocí in vivo videomikroskopie prokázala HPV dokonce i v arteriolách a venulách
o průměru pouhých 30-70 um, které mají jen minimální množství
hladkého svalu [26]. Japonská skupina Dr. Yamaguchi pomocí moderní techniky skanovací
konfokální laserové luminescenční mikroskopie v reálném čase in situ ukázala hypoxickou konstrikci plicních arteriol o průměru 20
- 30 um, zatímco průsvit podobně velkých venul ani průsvit kapilár
se při hypoxii nezměnil [94, 95].
In vitro se podařilo vyvolat specifickou konstrikci plicních arteriol
o průměru menším než 300 um (u kočky) závislou na stupni hypoxie.
Tepny větší než 500 um na hypoxii nereagovaly [27, 28]. V našich pokusech vyvolala akutní hypoxie konstrikci plicních
arteriol potkana o průměru ~300 um a menším, zatímco ve velkých
plicních tepnách (~1 mm) následovala po krátké přzvětšechodné
konstrikci setrvalá dilatace (Obr. 2-1) [45].
Nagasaka se spolupracovníky [29] měřili v izolovaných perfundovaných plicích koček tlak krve
v plícnici, v levé síni a pomocí mikropunkce také v arteriolách
a venulách o průměru 30-50 um. Zjistili, že při akutní hypoxii
se zvětšil rozdíl mezi všemi čtyřmi místy měření. Rozdíl mezi
plícnicí a arteriolami však vzrostl mnohem více než mezi tepénkami
a žilkami a mezi venulami a levou síní. To ukazuje, že se při
hypoxii nejvíce stahují arterioly větší než 30 um, ale jistý nevelký
podíl mají také menší arterioly a venuly a také žíly. Podobné
výsledky publikovali později s použitím obdobné metodiky Toga
a spol [93]. Dále to bylo potvrzeno také mikroskopickým pozorováním plicní
tkáně novorozených křečků transplantované do tvářového laloku
dospělých [30]. Akutní hypoxie vedla ke zúžení plicních arteriol a méně i plicních
venul, zatímco průměr tvářových cév se neměnil nebo lehce vzrostl.
K podobnému závěru - hlavní roli arteriálního a sekundární úloze
středního a žilního segmentu - dospěly také různé analýzy cévních
okluzí. Žilní okluze byla použita v izolovaném laloku psa [31] a v izolovaných plicích prasete [32]. Metoda arteriální okluze byla modifikována pro použití u intaktních
ovcí s obdobným výsledkem [33].
Hakim se spolupracovníky [34, 35] zjistili metodou arteriální a žilní okluze, že hlavním místem
HPV je střední poddajný segment plicního řečiště, i když k určité
konstrikci došlo i proximálně od tohoto úseku. Podobné výsledky
byly získány v nasí laboratoři [90] (Obr. 6-2). Střední segment zahrnuje kromě kapilár i venuly a arterioly
až do průměru cca 550 mm [36]. Doplnění metodiky o dvojitou okluzi (současně arteriální i
venózní) umožnilo dále rozdělit střední segment na prekapilární
arterioly a postkapilární venuly. Ukázalo se, že v perfundovaném
psím plicním laloku se při akutní hypoxii kontrahují nejvíce prekapilární
arterioly a jen do jisté míry i malé venuly a větší tepny [37, 92].
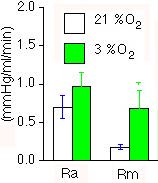
Obrázek 6-2: Akutní hypoxie zvyšuje hemodynamický odpor nejvíce ve středním
segmentu plicního oběhu (Rm) a poněkud méně v segmentu arteriálním
(Ra) [90]. Data jsou průměry +/- střední chyby průměru z pokusů využívajících
okluzní metody. Střední segment zahrnuje kromě kapilár i arterioly
a venuly.
Na základě nálezu HPV ve středním segmentu byly jako místo HPV navrženy i plicní kapiláry. Ty se však nemohou aktivně kontrahovat,
protože s výjimkou řídkých pericytů nemají buňky s kontraktilním
aparátem. Kapanci se spolupracovníky [38] zjistili, že proužky plicního parenchymu člověka, potkana a
krávy se in vitro kontrahují při snížení PO2. Mohl by to být důsledek stahu intersticiálních buněk v alveolárních
septech [39]. Nelze vyloučit možnost ovlivnění geometrie kapilárního řečiště
takovým stahem. Nález Kapanciho skupiny se však nepodařilo reprodukovat.
Proužky psího plicního parenchymu při hypoxii naopak relaxovaly
[40, 41]. Proti významné úloze kontrakce intersticia svědčí také skutečnost,
že akutní hypoxie zpravidla nemění plicní poddajnost ani odpor
dýchacích cest [42]. Ostatně úloha středního cévního segmentu byla, jak už bylo
uvedeno, uspokojivě objasněna tím, že zahrnuje i nejmenší arterioly
a venuly. V nedávné době byla však in vitro potvrzena schopnost intersticiálních buněk interalveolárních
sept kontrahovat se při hypoxii [91]. Zda takováto kontrakce může ovlivňovat plicní hemodynamiku
ovšem není vůbec jasné.
Hlavním místem HPV tedy jsou plicní tepny a arterioly, popřípadě také nejmenší venuly
[26]. Případný růst odporu v jiných cévních úsecích je při hypoxii
méně významný. Je možné, že se poněkud uplatňuje i hypoxií způsobený
pokles schopnosti erytrocytů se deformovat [43].
6.2.2 Depolarizace cévního hladkého svalu při HPV
Bergofsky a Holtzman [44] analyzovali obsah sodných a draselných iontů v izolovaných cévách
a v jejich kultivačním médiu po 30 minutách hypoxie. Zjistili,
že v hladkém svalu plicních arteriol ubylo K+ a přibylo Na+. Naměřené hodnoty dosadili do Nernstovy rovnice. Hypoxii odpovídala
depolarizace z -57 mV na -51 mV. Podobné změny nenastaly v plicních
žilách ani v systémových cévách.
Přímá měření membránového potencálu skleněnou intracelulární elektrodou
[27, 28] ukázala depolarizaci membrány plicní arterioly (průměr < 290
um) z -51 na -37 mV do 5 minut po snížení PO2 média ze 400 na 50 Torr. S pokračující hypoxií se začaly objevovat
stále častější akční potenciály. Větší plicní tepny na hypoxii
depolarizací ani tvorbou akčních potenciálů nereagovaly.Podobná
depolarizace buněk hladkého svalu plicních arteriol při hypoxii
byla prokázána také metodou terčíkového zámku ("patch clamp")
[45] (Obr. 6-3) a spektrofluorometricky (fluorescenční indikátor membránového
potenciálu DiBAC4) [46]. Myocyty systémových cév (které při hypoxii dilatují) odpovídají
na hypoxii hyperpolarizací [87].
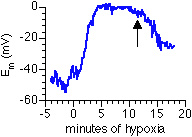
Obrázek 6-3: Akutní hypoxie depolarizuje buňku hladkého svalu izolovanou
z periferní plicní arterioly [45]. Membránový potenciál (Em) byl měřen metodou terčíkového zámku v konfiguraci proudového
zámku. Hypoxie byla zahájena v čase 0. Její ukončení je označeno
šipkou.
6.2.2.1 Inhibice draslíkových kanálů
Z iontových kanálů určují úroveň klidového membránového potenciálu
plicního cévního hladkého svalu nejvýrazněji kanály draslíkové,
zejména jejich podskupina řízená napětím [47, 45, 51]. Bylo opakovaně potvrzeno, že aktivita těchto kanálů v plicních
cévních myocytech klesá při hypoxii [48, 49, 50, 51, 45, 52, 88] (Obr. 6-4). Draslíkové kanály myocytů systémových cév jsou podle různých
autorů hypoxií buď aktivovány (což by mohlo být zčásti podkladem
vazodilatační odpovědi těchto cév na hypoxii), nebo nejsou signifikantně
ovlivněny.

- Obrázek 6-4: Hypoxie inhibuje draslíkový kanál sarkolemmy myocytu izolovaného
z plicní arterioly laboratorního potkana [45].
Obrázek ukazuje originální záznam proudu terčíkem mebrány obsahujícím
3 exempláře tohoto kanálu. Otevření každého z nich vedlo k jednotkovému
zvýšení proudu na další vyšší hodnotu naznačenou značkami uplně
vpravo (C značí všechny 3 kanály zavřené). Pravděpodobnost otevření
kanálu byla signifikantně a reverzibilně snížena při hypoxii.
Co vede ke snížení aktivity draslíkových kanálů buněk hladkého
svalu plicních cév při hypoxii není přesně zřejmé. Předpokládá
se, že kanály nebo některé jejich podjednotky mohou být přímo
regulovány redoxním stavem buňky. Je také možné, že se mohou uplatňovat změny intracelulárního
pH. Intracelulární acidifikace, ke které v hladkém svalu plicních
cév při hypoxii dochází (Madden), inhibuje napětím řízené draslíkové
kanály [96]. V systémových cévách jsou tyto kanály poklesem intracelulárního
pH naopak aktivovány [96]. Tento rozdíl by mohl být podkladem opačné odpovědi plicních
a systémových cév na hypoxii.
Funkční význam této inhibice ukazují pokusy, v nichž farmakologické
inhibitory draslíkových kanálů, zejména kanálů řízených napětím,
napodobily HPV v izolovaných perfundovaných plicích či izolovaných arteriolách
[48, 45]. Podobný účinek má zvýšení extracelulární koncentrace K+. Umělé zvýšení extracelulární koncentrace draslíku snižuje rozdíl
koncentrace K+ mezi buňkou a jejím okolím. To působí ustavení nové rovnováhy,
jejímž důsledkem je méně negativní potenciál vnitřní strany membrány,
tedy depolarizace. Takový zákrok vede v izolovaných plicích k
posílení reaktivity na různé podněty včetně hypoxie [44, 53].
Depolarizace při hypoxii zřejmě působí vazokonstrikci prostřednictvím
aktivace vápníkových kanálů řízených napětím. Je ale zajímavé, že tvorba akčních potenciálů
a depolarizace jsou inhibovány blokátorem těchto kanálů verapamilem
[27]. Je tedy možné, že se při HPV uplatňuje pozitivní zpětná vazba, v níž depolarizace vede ke
vstupu Ca2+ do hladkého svalu aktivovanými Ca2+ kanály, což dále podporuje depolarizaci. Navíc je možné, že inhibice
draslíkových kanálů při hypoxii může být sekundárním důsledkem
uvolnění Ca2+ ze sarkoplasmatického retikula. Inhibici draslíkových kanálů
silnou hypoxií totiž může zabránit chelace intracelulárního Ca2+ nebo farmakologická blokáda uvolnění Ca2+ z intracelulárních zdrojů [51]. Zvýšení [Ca2+]i v plicních cévních myocytech při hypoxii časově předchází depolarizaci
[51]. Změny [Ca2+]i při HPV jsou diskutovány níže.
Na polarizaci membrány se může podílet elektrogenní Na/K-ATPáza
[54]. Její inhibice tedy napomáhá depolarizaci. Při HPV se však takový mechanismus asi neuplatňuje. Inhibice Na/K pumpy
totiž HPV neposiluje, ale naopak oslabuje [55]. Membránová Na/K-ATPáza tedy pravděpodobně nemusí v hladkém
svalu plicních cév být elektrogenní (obdobně jako v řadě jiných
cév).
Kontraktilní aparát cévního hladkého svalu obsahuje myosin, myosinkinázu,
kalmodulin a komplex aktin-tropomyosin-kaldesmon (Obr. 6-5). Aktivaci tohoto systému zahajuje navázání ionizovaného vápníku
na kaldesmon. Ten se tím uvolní od komplexu aktin-tropomyosin
a spolu s vápenatým iontem se připojí ke kalmodulinu. Tím se odblokuje
schopnost aktinového komplexu interagovat s myosinem a současně
získá komplex kaldesmon-kalmodulin- Ca2+ schopnost aktivovat myosinkinázu B. Fosforylace lehkého řetězce
myosinu myosinkinázou pak aktivuje ATPázovou funkci myosinu i
jeho interakci s aktinem, která je podstatou kontrakce [56, 57, 58].
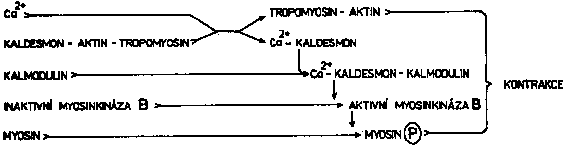
- Obrázek 6-5: Schéma funkce kontraktilního aparátu cévního hladkého svalu.
- Fosforylaci myosinu navíc může pomáhat udržovat myosinkináza C,
aktivovaná diacylglycerolem vzniklým stimulací fosfolipázy C.
Podle McCalla [56], Reutera [57], Rasmussena a spolupracovníků [59] a Karakiho a Weisse [58].
Intracelulární koncentrace Ca2+ je rovnováhou mezi vstupem vápníku do cytoplazmy a jeho odčerpáváním
z ní. Ca2+ se dostává do cytoplazmy z extracelulárního prostoru specifickými
kanály a (jen minimálně) difúzí, anebo se do ní uvolňuje zejména
ze sarkoplazmatického retikula (uvolňování z mitochondrií a plazmalemmy
je za fyziologických podmínek prakticky nevýznamné) [60]. Odčerpává se aktivně buď ven z buňky, anebo do sarkoplasmatického
retikula. Depolarizace plasmatické membrány aktivuje napětím řízené
Ca2+ kanály (v cévním hladkém svalu především typu L) [61]. Aktivace receptorů různých agonistů zvyšuje intracelulární
koncentraci Ca2+ jednak otevřením specifického kanálu řízeného příslušným receptorem,
a navíc aktivuje membránovou fosfolipázu C. Ta štěpí fosfatidylinositol
bisfosfát z buněčné membrány na inositoltrifosfát, který otevřením
specifického kanálu uvolňuje Ca2+ ze sarkoplazmatického retikula, a na diacylglycerol, který navíc
aktivuje myosinkinázu C. Ta napomáhá udržovat kontrakci [62, 59].
Z výše uvedeného je zřejmé, ze součástí HPV musí být buď zvýšení [Ca2+]i v kompartmentu kontraktilního aparátu, anebo zvýšení citlivosti
tohoto aparátu k Ca2+. Řada nálezů dokazuje, ze součástí mechanismu HPV je zvýšení [Ca2+]i v plicních cévních myocytech (i když existují i údaje svědčící
pro zvýšenou citlivost kontraktilního aparátu k Ca2+ [63]). V izolovaných plicích nelze vyvolat HPV, odstraní-li se z perfuzátu Ca2+ [64]. Hypokalcémie in vivo ruší HPV [65]. Hypoxickou plicní vazokonstrikci naopak posiluje ortovanadát,
který (poměrně neselektivně) inhibuje Ca2+ ATPázu odčerpávající vápník z cytoplasmy [64]. Síla hypoxických kontrakcí izolovaných plicních arteriol in vitro závisí na koncentraci Ca2+ v médiu [27, 66]. Mg2+, přirozený antagonista vápníkových kanálů [67], inhibuje HPV [68]. Zvýšení [Ca2+]i při hypoxii bylo pomocí spektrofluoroskopického indikátoru fura
2 přímo změřeno v myocytech malých plicních cév [69, 70, 71, 46] a v izolovaných periferních plicních arteriolách [63]. Tato odpověď je výrazně oslabená [71] nebo ji vůbec nelze vyvolat v nepřítomnosti extracelulárního
Ca2+ [69, 70].
6.2.3.1 Aktivace vápníkových kanálů plasmalemmy
Vzhledem k přítomnosti deoplarizace při HPV nepřekvapuje, že aktivace L-typu Ca2+ kanálu (řízeného napětím) je velmi významným mechanismem influxu
Ca2+ při HPV. Blokátory těchto kanálů (verapamil, nifedipin, nitrendipin,
SKF 525 A) konzistentně inhibují plicní vazokonstrikční odpověď
na hypoxii [72, 73, 74, 75, 76, 77, 27, 78, 79, 80, 81, 65]. Naopak látka BAY K 8644, která facilituje průchod vápenatých
iontu částecně aktivovanými pomalými vápníkovými kanály řízenými
napětím, HPV potencuje [82, 80]. Stejný účinek nemá látka A23187, která zvyšuje intracelulární
koncentraci Ca2+ nezávisle na iontových kanálech [82]. Účinek vápníkových "blokátorů" je specifický pro HPV v tom smyslu, ze reaktivitu na jiné podněty (angiotensin II a
prostaglandin F2alfa) tlumí jen mírně nebo vůbec [72]. To pravděpodobně souvisí s úlohou kanálů řízených příslušnými
receptory (viz výše). Inhibitory L-typu Ca2+ kanálu redukují také hypoxický vzrůst [Ca2+]i v plicních cévních myocytech [70, 46].
Aktivace Ca2+ kanálů typu L hypoxií byla v myocytech malých plicních arteriol
nedávno přímo změřena metodou terčíkového zámku [52]. Ca2+ kanály myocytů velkých plicních cév (které se při hypoxii nekontrahují)
takovou odpověď nevykazovaly. Ca2+ kanály myocytů systémových cév (které při hypoxii dilatují) byly
hypoxií inhibovány [83]. Tyto odpovědi byly naměřeny i při použití napěťového zámku,
takže aktivace Ca2+ kanálů nebyla v tomto případě zprostředkována depolarizací. Autoři
se domnívají, ze šlo o přímé působení hypoxie na Ca2+ kanály anebo s nimi přímo spřažený protein. V každém případě
je nejvýš pravděpodobné, že in vivo bude takováto odpověď dále potencována depolarizací.
6.2.3.2 Úloha sarkolasmatického retikula
Uvolnění Ca2+ ze sarkoplasmatického retikula se při HPV pravděpodobně také uplatňuje, i když jeho přesná úloha není jasná.
Deplece Ca2+ ze sarkoplazmatického retikula thapsigarginem zabraňuje hypoxickému
zvýšení [Ca2+]i v plicních cévních myocytech [70] a zmenšuje HPV v izolovaných plicích [84]. Obě tyto odpovědi jsou také redukovány inhibitory Ca2+ kanálů sarkoplasmatického retikula ryanodinem a kofeinem [70, 84, 46].
Existují protichůdné názory na to, zda je uvolnění Ca2+ z endoplasmatického retikula při HPV příčinou nebo následkem influxu Ca2+ přes plasmatickou membránu. Cornfield a spol. [46] zjistili, že v přítomnosti ryanodinu zvýšila hypoxie [Ca2+]i v plicních cévních myocytech pouze přechodně. To naznačuje, že
uvolnění Ca2+ z retikula je důsledkem influxu Ca2+ (tzv. kalcium indukované uvolnění kalcia, CICR [60]). Naproti tomu Salvaterrová a Goldman [70] zjistili, že hypoxické zvýšení [Ca2+]i bylo nejen redukováno, ale i výrazně zpomaleno kofeinem. To by
svědčilo pro důležitou úlohu uvolnění Ca2+ z retikula pro nastartování hypoxické odpovědi. Tuto možnost,
která připomíná stále ještě poněkud elusivní fenomén tzv. kapacitního
influxu Ca2+ [85, 60], podporuje nepřímo i údaj, podle něhož hypoxické zvýšení [Ca2+]i časově předchází inhibici draslíkových kanálů, jíž lze předejít podáním kofeinu [51].
Kromě Ca2+ kanálů se v HPV snad může uplatňovat i systém výměny sodíku za vápník. Schopnost
tohoto systému odstraňovat Ca2+ z cytoplazmy je při hypoxii snížená [86]. Zmenšení sodíkového gradientu totiž může omezit Na/Ca výměnu.
LITERATURA
1. Bland RD, Demling RH, Selinger SL, Staub NC: Effects of alveolar
hypoxia on lung fluid and protein transport in unanesthetized
sheep. Circ Res 1976; 40: 269-274.
2. Aarseth P, Karlsen J: Blood volume and extravascular water
content in the rat lung during acute alveolar hypoxia. Acta Physiol
Scand 1977; 100: 236-245.
3. Cheney FW, Bishop MJ, Eisenstein BL, Artman LD: Hypoxic pulmonary
vasoconstriction does not affect hydrostatic pulmonary edema formation.
J Appl Physiol 1987; 62: 776-780.
4. Sackner MA, Will DH, Dubois AB: The site of pulmonary vasomotor
activity during hypoxia or serotonin administration. J Clin Invest
1966; 45: 112-120.
5. Glazier JB, Murray JF: Sites of pulmonary vasomotor reactivity
in the dog during alveolar hypoxia and serotonin and histamine
infusion. J Clin Invest 1971; 50: 2550-2558.
6. Quebbeman EJ, Dawson CA: Effect of lung inflation and hypoxia
on pulmonary arterial blood volume. J Appl Physiol 1977; 43: 8-13.
7. Dawson CA, Jones RL, Hamilton LH: A pulmonary circulation model
based on forward and retrograde perfusion of isolated lungs. J
Appl Physiol 1973; 35: 103-110.
8. Aarseth P, Bjertnaes L, Karlsen J: Changes in blood volume
and extravascular water content in isolated perfused rat lungs
during ventilation hypoxia. Acta Physiol Scand 1980; 109: 61-67.
9. DeLeon SR, Orchard CH, Chakrabarti MK, Sykes MK: Effect of
hypoxia on fluid filtration rate during forward and reverse perfusion
of isolated rabbit lungs. Cardiovasc Res 1982; 16: 711-715.
10. Kuida H, Brown AM, Thorne JL, Lange RL, Hecht HH: Pulmonary
vascular response to acute hypoxia in normal, unanesthetized calves.
Am J Physiol 1962; 203: 391-396.
11. Lloyd T: Effect of alveolar hypoxia on pulmonary vascular
resistance. J Appl Physiol 1964; 19: 1086-1094.
12. Morkin E, Levine OR, Fishman AP: Pulmonary capilarry flow
pulse and the site of pulmonary vasoconstriction in the dog. Circ
Res 1964; 15: 146-160.
13. Malik AB, Kidd BS: Pulmonary arterial wedge and left atrial
pressures and the site of hypoxic pulmonary vasoconstriction.
Respiration 1976; 33: 123-132.
14. Doyle JT, Wilson JS, Warren JV: The pulmonary vascular responses
to short-term hypoxia in human subjects. Circulation 1952; 5:
263-270.
15. Robin ED, Theodore J, Burke CM, Oesterle SN, Fowler MB, Jamieson
SW, Baldwin JC, Morris AJ, Hunt SA: Hypoxic pulmonary vasoconstriction
persists in the human transplanted lung. Clin Sci 1987; 72: 283-287.
16. Guazzi MD, Berti M, Doria E, Fiorentini C, Galli C, Pepi M,
Tamborini G: Enhancement of the pulmonary vasoconstriction reaction
to alveolar hypoxia in systemic high blood pressure. Clin Sci
1989; 76: 589-594.
17. Herles F: The pulmonary artery wedge pressure. Cor Vasa 1966;
8: 161-166.
18. Reeves JT, Groves BM: Approach to the patient with pulmonary
hypertension. In: Weir EK, Reeves JT, Eds. Pulmonary Hypertension.
Mount Kisco, Futura, 1984; 1-44.
19. Aviado DM: Pulmonary venular responses to anoxia, 5-hydroxytriptamine
and histamine. Am J Physiol 1960; 198: 1032-1036.
20. Hyman AL, Kadowitz PJ: Effects of alveolar and perfusion hypoxia
and hypercapnia on pulmonary vascular resistance in the lamb.
Am J Physiol 1975; 228: 397-403.
21. Hirschman JC, Bouncek RJ: Angiographic evidence of pulmonary
vasomotion in the dog. Br Heart J 1963; 25: 375-381.
22. Allison DJ, Stanbrook HS: A radiologic and physiologic investigation
into hypoxic pulmonary vasoconstriction in the dog. Invest Radiol
1980; 15: 178-190.
23. Kato M, Staub NC: Response of small pulmonary arteries to
unilobar hypoxia and hypercapnia. Circ Res 1966; 19: 426-439.
24. Koyama T, Horimoto M: Blood flow reduction in local pulmonary
microvessels during acute hypoxia imposed on a small fraction
of the lung. Respir Physiol 1983; 52: 181-189.
25. Shirai M, Sada K, Ninomiya I: Effects of regional alveolar
hypoxia and hypercapnia on small pulmonary vessels in cats. J
Appl Physiol 1986; 61: 440-448.
26. Hillier SC, Graham J, Hanger C, Godbey PS, Glenny RW, Wagner
WW: Hypoxic vasoconstriction in pulmonary arterioles and venules.
J Appl Physiol 1997; 82: 1084-1090.
27. Harder DR, Madden JA, Dawson C: Hypoxic induction of Ca2+-dependent
action potentials in small pulmonary arteries of the cat. J Appl
Physiol 1985; 59: 1389-1393.
28. Madden JA, Dawson CA, Harder DR: Hypoxia-induced activation
in small isolated pulmonary arteries from the cat. J Appl Physiol
1985; 59: 113-118.
29. Nagasaka K, Bhattacharya J, Nanjo S, Gropper MA, Staub NC:
Micropuncture measurement of lung microvascular pressure profile
during hypoxia in cats. Circ Res 1984; 54: 90-95.
30. Shepard JM, Joyner WL, Gilmore JP: Hypoxia does not alter
angiotensin converting enzyme activity in hamster pulmonary microvessels.
Circ Res 1987; 61: 228-235.
31. Dawson CA, Linehan JH, Bronikowski TA, Rickaby DA: Pulmonary
microvascular hemodynamics: occlusion methods. In: Will JA, Dawson
CA, Weir EK, Buckner CK, Eds. The Pulmonary Circulation in Health
and Disease. Orlando, Academic Press, 1987; 175-197.
32. Rock P, Patterson GA, Permutt S, Sylvester JT: Nature and
distribution of vascular resistance in hypoxic pig lungs. J Appl
Physiol 1985; 59: 1891-1901.
33. Siegel LC, Pearl RG, Shafer SL, Ream AK, Prielipp RC: The
longitudinal distribution of pulmonary vascular resistance during
unilateral hypoxia. Anesthesiology 1989; 70: 527-532.
34. Hakim TS, Michel RP, Minami H, Chang HK: Site of pulmonary
hypoxic vasoconstriction studied with arterial and venous occlusion.
J Appl Physiol 1983; 54: 1298-1302.
35. Hakim TS, Malik AB: Hypoxic vasoconstriction in blood and
plasma perfused lungs. Respir Physiol 1988; 72: 109-122.
36. Tod M, McGeady ML, Rock P, Sylvester JT: Effect of arterial
ligation and embolization on pulmonary vascular pressure distribution.
J Appl Physiol 1987; 63: 1387-1395.
37. Hakim TS: Identification of constriction in large versus small
vessels using the arterial-venous and the double-occlusion techniques
in isolated canine lungs. Respiration 1988; 54: 61-69.
38. Kapanci Y, Assimacopoulas A, Zwahlen A, Gabbiani G: Contractile
interstitial cells in pulmonary alveolar septae: a possible regulator
of ventilation/perfusion ratio? J Cell Biol 1974; 60: 375-392.
39. Mazzone R: Effect of hypoxia on alveolar capillary morphology.
Physiologist 1980; 23: 154.
40. Duane SF, Weir EK, Stewart RM, Niewoehner DE: Distal airway
responses to changes in oxygen and carbon dioxide tensions. Respir
Physiol 1979; 38: 303-311.
41. Stewart RM, Weir EK, Montgomery MR, Niewoehner DE: Hydrogen
peroxide contracts airway smooth muscle: a possible endogenous
mechanism. Respir Physiol 1981; 45: 333-342.
42. Holroyde MC: Do leukotrienes mediate hypoxic pulmonary vasoconstriction
(HPV) in dogs? Br. J. Pharmacol. 1986; 88: 395P.
43. Hakim TS, Macek AS: Role of erythrocyte deformability in the
acute hypoxic pressor response in the pulmonary vasculature. Respir
Physiol 1988; 72: 95-98.
44. Bergofsky EH, Holtzman S: A study of the mechanisms involved
in the pulmonary arterial pressor response to hypoxia. Circ Res
1967; 20: 506-519.
45. Archer SL, Huang JMC, Reeve HL, Hampl V, Tolarová S, Michelakis
E, Weir EK: Differential distribution of electrophysiologically
distinct myocytes in conduit and resistance arteries determines
their response to nitric oxide and hypoxia. Circ Res 1996; 78:
431-442. (abstrakt)
46. Cornfield DN, Stevens T, McMurtry IF, Abman SH, Rodman DM:
Acute hypoxia causes membrane depolarization and calcium influx
in fetal pulmonary artery smooth muscle cells. Am J Physiol 1994;
266: L469-L475.
47. Yuan X-J: Voltage gated K+ currents regulate resting membrane
potential and [Ca2+]i in pulmonary artery myocytes. Circ Res 1995;
77: 370-378.
48. Post JM, Hume JR, Archer SL, Weir EK: Direct role for potassium
channel inhibition in hypoxic pulmonary vasoconstriction. Am J
Physiol 1992; 262: C882-C890.
49. Archer SL, Huang J, Henry T, Peterson D, Weir EK: A redox-based
O2 sensor in rat pulmonary vasculature. Circ Res 1993; 73: 1100-1112.
50. Yuan X-J, Goldman WF, Tod ML, Rubin LJ, Blaustein MP: Hypoxia
reduces potassium currents in cultured rat pulmonary but not mesenteric
arterial myocytes. Am J Physiol 1993; 264: L116-L123.
51. Post JM, Gelband CH, Hume JR: [Ca2+]i inhibition of K+ channels
in canine pulmonary artery: novel mechanism for hypoxia-induced
membrane depolarization. Circ Res 1995; 77: 131-139.
52. Franco-Obregón A, López-Barneo J: Differential oxygen sensitivity
of calcium channels in rabbit smooth muscle cells of conduit and
resistance pulmonary arteries. J Physiol 1996; 491: 511-518.
53. Hauge A: Ventilation hypoxia and pulmonary vascular resistance:
effects of changes in plasma potassium concentration. Acta Physiol
Scand 1969; 75: 240-244.
54. Skou JC: Overview: The Na,K-pump. Methods Enzymol 1988; 156:
1-25.
55. Herget J, McMurtry IF: Effect of ouabain, low K+, and aldosterone
on hypoxic pressor reactivity of rat lungs. Am J Physiol 1985;
248: H55-H60.
56. McCall D: Excitation-contraction coupling in cardiac and vascular
smooth muscle: modification by calcium-entry blockade. Circulation
1987; 75: 3-14.
57. Reuter H: Modulation of ion channels by phosphorylation and
second messengers. News Physiol Sci 1987; 2: 168-171.
58. Karaki H, Weiss GB: Calcium release in smooth muscle. Life
Sci 1988; 42: 111-122.
59. Rasmussen H, Takuwa Y, Park S: Protein kinase C in the regulation
of smooth muscle contraction. FASEB J 1987; 1: 177-185.
60. Pozzan T, Rizzuto R, Volpe P, Meldolesi J: Molecular and cellular
physiology of intracellular calcium stores. Physiol Rev 1994;
74: 595-636.
61. McDonald TF, Pelzer S, Trautwein W, Pelzer DJ: Regulation
and modulation of calcium channels in cardiac, skeletal, and smooth
muscle cells. Physiol Rev 1994; 74: 366-507.
62. Casteels R, Droogmans G: Cell membrane responsiveness and
excitation-contraction coupling in smooth muscle. J Cardiovasc
Pharmacol 1984; 6: S304-S312.
63. Robertson TP, Aaronson PI, Ward JPT: Hypoxic intracellular
Ca2+ in pulmonary arteries: evidence for PKC-independent Ca2+
sensitization. Am J Physiol 1995; 37: H301-H307.
64. Farrukh IS, Gurtner GH, Michael JR: Effect of inhibiting calcium
ATPase on pulmonary artery pressure during normoxia and hypoxia.
Am. Rev. Respir. Dis. 1986; 133: A226.
65. Drop LJ, Toal KW, Geffin GA, O'Keefe DD, Hoaglin DC, Daggett
WM: Pulmonary vascular responses to hypercalcemia and hypocalcemia
in the dog. Anesthesiology 1989; 70: 825-836.
66. Yuan X-J, Tod ML, Rubin LJ, Blaustein MP: Contrasting effects
of hypoxia on tension in rat pulmonary and mesenteric arteries.
Am J Physiol 1990; 259: H281-H289.
67. Altura BM, Altura BT, Carella A, Gebrewold A, Marakawa T,
Nishio A: Mg2+ - Ca2+ interaction in contractility of vascular
smooth muscle: Mg2+ versus organic calcium channel blockers on
myogenic tone and agonist-induced responsiveness of blood vessels.
Can J Physiol Pharmacol 1987; 65: 729-745.
68. Cropp GJA: Reduction of hypoxic pulmonary vasoconstriction
by magnesium chloride. J Appl Physiol 1968; 24: 755-760.
69. Cornfield DN, Stevens T, McMurtry IF, Abman SH, Rodman DM:
Acute hypoxia increases cytosolic calcium in fetal pulmonary artery
smooth muscle cells. Am J Physiol 1993; 265: L53-L56.
70. Salvaterra CG, Goldman W: Acute hypoxia increases cytosolic
calcium in cultured pulmonary arterial myocytes. Am J Physiol
1993; 264: L323-L328.
71. Vadula MS, Kleinman JG, Madden JA: Effect of hypoxia and norepinephrine
on cytoplasmic free Ca2+ in pulmonary and cerebral arterial myocytes.
Am J Physiol 1993; 265: L591-L597.
72. McMurtry IF, Davidson AB, Reeves JT, Grover RF: Inhibition
of hypoxic pulmonary vasoconstriction by calcium antagonists in
isolated rat lungs. Circ Res 1976; 38: 99-104.
73. Tucker A, McMurtry IF, Grover RF, Reeves JT: Attenuation of
hypoxic pulmonary vasoconstriction by verapamil in intact dogs.
Proc Soc Exp Biol Med 1976; 151: 611-614.
74. Suggett AJ, Mohammed FH, Barer GR: Angiotensin, hypoxia, verapamil
and pulmonary vessels. Clin Exp Pharmacol Physiol 1980; 7: 263-274.
75. Bishop MJ, Cheney FW: Comparison of the effects of minoxidil
and nifedipine on hypoxic pulmonary vasoconstriction in dogs.
J Cardiovasc Pharmacol 1983; 5: 184-189.
76. Young TE, Lundquist LJ, Chesler E, Weir EK: Comparative effects
of nifedipine, verapamil, and diltiazem on experimental pulmonary
hypertension. Am J Cardiol 1983; 51: 195-200.
77. Stanbrook HS, Morris KG, McMurtry IF: Prevention and reversal
of hypoxic pulmonary hypertension by calcium antagonists. Am Rev
Respir Dis 1984; 130: 81-85.
78. Philips JB, Lyrene RK, Leslie GI, McDevitt M, Cassady G: Hemodynamic
effects of nifedipine in normoxic and hypoxic newborn lambs. Pediatr
Pharmacol 1985; 5: 23-30.
79. Escourrou P, Simonneau G, Ansquer JC, Duroux P, Lockhart A:
A single orally administred dose of almitrine improves pulmonary
gas exchange during exercise in patients with chronic air-flow
obstruction. Am Rev Respir Dis 1986; 133: 562-568.
80. Tolins M, Weir EK, Chesler E, Nelson DP, From AHL: Pulmonary
vascular tone is increased by a voltage-dependent calcium channel
potentiator. J Appl Physiol 1986; 60: 942-948.
81. Mélot C, Naeije R, Hallemans R, Lejeune P, Mols P: Hypoxic
pulmonary vasoconstriction and pulmonary gas exchange in normal
man. Respir Physiol 1987; 68: 11-27.
82. McMurtry IF: BAY K 8644 potentiates and A23187 inhibits hypoxic
vasoconstriction in rat lungs. Am J Physiol 1985; 249: H741-H746.
83. Franco-Obregón A, UreEa J, López-Barneo J: Oxygen-sensitive
calcium channels in vascular smooth muscle and their possible
role in hypoxic arterial relaxation. Proc Natl Acad Sci USA 1995;
92: 4715-4719.
84. Archer SL, Hampl V, Nelson D, Weir EK: Inhibitors of sarcoplasmic
reticulum release and reuptake attenuate several types of pulmonary
vasoconstriction in isolated rat lungs. Circulation 1994; 90:
I-151.
85. Hoth M, Penner R: Depletion of intracellular calcium stores
activates a calcium current in mast cells. Nature 1992; 355: 353-356.
86. Salvaterra CG, Rubin LJ, Schaeffer J, Blaustein MP: The influence
of the transmembrane sodium gradient on the responses of pulmonary
arteries to decreases in oxygen tension. Am Rev Respir Dis 1989;
139: 933-939.
87. Lombard JH, Smeda J, Madden JA, Harder DR: Effect of reduced
oxygen availability upon myogenic depolarization and contraction
of the cat middle cerebral artery. Circ Res 1986; 58: 565-569.
88. Osipenko ON, Evans AM, Gurney AM: Regulation of the resting
potential of rabbit pulmonary artery myocytes by a low threshold,
O2-sensing potassium current. Br J Pharmacol 1997; 120: 1461-1470.
89. Chammas JH, Rickaby DA, Guarin M, Linehan JH, Hanger CC, Dawson
CA: Flow-induced vasodilation in the ferret lung. J Appl Physiol
1997; 83: 495-502.
90. Herget J, Kuklik V: Perinatal lung injury extends in adults
the site of hypoxic pulmonary vasoconstriction upstream. Physiol
Res 1995; 44: 25-30.
91. Fukui M, Yasui H, Watanabe K, Fujimoto T, Kakuma T, Yoshida
R, Ohi M, Kuno K: Hypoxic contraction of contractile interstitial
cells isolated from bovine lung. Am J Physiol 1996; 14: L962-L972.
92. Ferrario L, Amin HM, Sugimori K, Camporesi EM, Hakim TS: Site
of action of endogenous nitric oxide on pulmonary vasculature
in rats. Pflugers Arch 1996; 432: 523-527.
93. Toga H, Okazaki H, Ishigaki M, Noguchi T, Huang J, Fukunaga
T, Nagasaka Y, Takahashi K, Ohya N: Effect of hypoxia on pulmonary
blood flow- segmental vascular resistance relationship in perfused
cat lungs. J Appl Physiol 1998; 84: 1003-1010.
94. Yamaguchi K, Suzuki K, Naoki K, Nishio K, Sato N, Takeshita
K, Kudo H, Aoki T, Suzuki Y: Response of intra-acinar pulmonary
microvessels to hypoxia, hypercapnic acidosis, and isocapnic acidosis.
Circ Res 1998; 82: 722-728.
95. Suzuki K, Naoki K, Kudo H, Nishio K, Sato N, Aoki T, Suzuki
Y, Takeshita K, Miyata A: Impaired hypoxic vasoconstriction in
intraacinar microvasculature in hyperoxia-exposed rat lungs. Am
J Respir Crit Care Med 1998; 158: 602-609.
96. Berger MG, Vandier C, Bonnet P, Jackson WF, Rusch NJ: Intracellular
acidosis differentially regulates KV channels in coronary and pulmonary vascular muscle. Am J Physiol
1998; 275: H1351-H1359.