Jedním ze substrátů NO syntázy je molekulární kyslík [48]. Dostatečné snížení jeho dostupnosti proto snižuje syntézu NO in vitro [187]. To vedlo k atraktivní hypotéze, že mechanismem HPV může být hypoxické snížení plicní syntézy NO. Tuto hypotézu podpořily studie, které zjistily, že akutní hypoxie snižuje reaktivitu plicních tepen in vitro na vazodilatační látky, jejichž působení je zprostředkováno uvolněním NO z endotelu [188, 189, 190, 191]. To naznačilo sníženou schopnost endotelu tvořit v hypoxii NO. Nevýhodou tohoto typu pokusu je ovšem možnost, že hypoxie ovlivňuje vazbu ligandu s receptorem či intracelulární přenos signálu spíše než NO syntázu jako takovou. Vazba vazodilatační látky s jejím receptorem ovšem zjevně nemá nic společného s mechanismem HPV. Navíc postupně začalo přibývat údajů svědčících proti signifikantní bazální syntéze NO v plicních cévách (kap. 4.5). Rozhodli jsme se proto tuto otázku prostudovat jinými metodami.
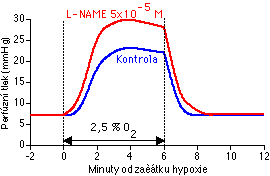
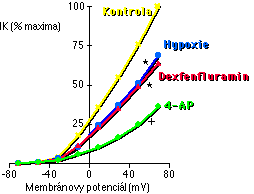
Nejprve jsme chtěli potvrdit v té době nové pozorování, že akutní podání inhibitorů NO syntázy potencuje HPV [89]. Náš identický výsledek (Obrázek 5-1) byl publikován v předběžné formě v roce 1992 [192] a posléze i v definitivní podobě [119]. Už v době naši práce na tomto tématu ovšem začalo přibývat publikací potvrzujících, že inhibitory NO syntázy potencují HPV [149, 90, 110, 91, 193, 194, 195, 113, 196, 114, 117, 139, 118, 146, 147, 151, 197]. My jsme navíc mezi tím ukázali, že HPV je značně potencována i chronickou aplikací inhibitorů NO syntázy.
Takový stupeň alveolární hypoxie, který je relevantním stimulem pro HPV in vivo (je ho možno prežít [198]), tj. cca 35 mmHg, tedy zjevně nejen že syntézu NO v plicních cévách neinhibuje, ale naopak spíše stimuluje. Nejpravděpodobnějším vysvětlením je, že afinita NO syntázy ke kyslíku je taková, že se kyslík stává limitujícím faktorem syntézy NO teprve až při velmi silné hypoxii. Naproti tomu při mírnějších stupních hypoxie, jaké jsou relevantní z hlediska regulace plicního cévního tonu in vivo, je NO syntáza zřejme pod rozhodujícím vlivem jiných regulačních mechanismů, jako např. intracelulární koncentrace volného vápenatého iontu ([Ca2+]i) [76, 77, 78, 49, 79].
Nevýhodou výše popsaných pokusů s inhibitory NO syntázy je možnost nespecifických účinků (kap. 3.2). Proto jsme se rozhodli měřit tvorbu NO plicním endotelem při akutní hypoxii přímo. Zajímalo nás také, zda podnětem pro změnu tvorby NO při HPV může být přímo hypoxie per se, bez ohledu na její hemodynamické důsledky v plicích (tj. zvýšené sřrižné napětí na endoteliálních buňkách v důsledku rychlejšího průtoku kontrahovanými cévami [100, 146, 199, 102, 200, 201, 202]).
Měřili jsme proto množství NO a jeho oxidačního produktu, NO2-, uvolněných do superfuzátu kultury hovězích plicních endoteliálních buněk v průběhu desetiminutové expozice hypoxii. Protože podnětem pro uvolňování NO z endotelu je přechodné zvýšení [Ca2+]i [76, 77, 78, 49, 79], testovali jsme také hypotézu, že akutní hypoxie vede k přechodnému zvýšení [Ca2+]i v plicních endoteliálních buňkách [125].
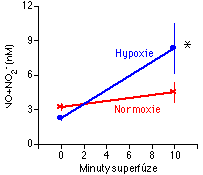
Zjistili jsme, že zatímco superfúze endoteliální kultury normoxickým roztokem signifikantně nezměnila koncentraci NO+NO2- v superfuzátu (P>0.05), identický pokus s hypoxickým superfuzátem vedl ke značné akumulaci NO+ NO2- v roztoku (Obrázek 5-2) [125, 126]. Byly-li ze systému vynechány buňky a prřdán exogenní NO, byly po 10 minutách hladiny NO+ NO2- shodné v normoxickém a hypoxickém roztoku. To ukazuje, že zvýšená akumulace NO+ NO2- v pokuse s buňkami nebyla důsledkem odlišné degradace NO za hypoxických podmínek.
Změny [Ca2+]i při akutní hypoxii jsme v plicních endoteliálních buňkách studovali pomoci fluorescenčního kalciového indikátoru fura-2 [203]. Diskuse naší implementace metody pro plicní endotel byla podrobně publikována [61]. Ve stručnosti lze shrnout, že hovězí plicní endoteliální buňky byly kultivovány v plastikových miskách, jejichž dno bylo zčásti nahrazeno podložním sklíčkem. To umožnilo osvit buněk potřebnými vlnovými délkami (340 a 380 nm) prostředníctvím invertovaného mikroskopu, na jehož temperovaném stolku byla miska během pokusu perfundována Hanksovým solným roztokem. Roztok byl před vstupem do misky probubláván vzduchem s 5% CO2 (výsledné PO2 =152 mmHg) anebo (po dobu 3 - 5 minut) dusíkem s 5% CO2 (výsledné PO2 = 37 mmHg).
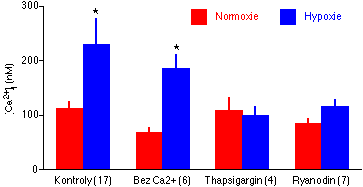
Zjistili jsme [125], že akutní hypoxie vyvolává přechodné (~40 s) zvýšení [Ca2+]i ze 113 +/- 10 nM na 231 +/- 45 nM (P<0.05, n = 17 misek). Změna z hypoxie zpět na normoxii k žádné změně [Ca2+]i nevedla. Hypoxické zvýšení [Ca2+]i nebylo zmenšeno vynecháním Ca2+ ze superfúzního roztoku, které snížilo normoxickou bazální úroveň [Ca2+]i i odpověď na bradykinin. Hypoxický vzrůst [Ca2+]i byl výrazně snížen přítomností ryanodinu a zcela eliminován thapsigarginem (Obrázek 5-3). Ryanodin působí depleci Ca2+ z endoplasmatického retikula tím, že "zmrazuje" jeden ze dvou typů kalciových kanálů v membráně endoplasmatického retikula v otevřeném stavu [204]. Thapsigragin vysoce selektivně inhibuje vápníkovou pumpu v membráně endoplasmatického retikula, která přečerpává Ca2+ z cytosolu do endoplasmatického retikula [205]. Inhibice této pumpy vede velmi rychle k depleci Ca2+ z endoplasmatického retikula [205, 206]. Podle současných představ je endoplasmatické retikulum jedinou intracelulární zásobarnou Ca2+ významnou pro regulaci [Ca2+]i za fyziologických podmínek (pro podrobný přehled a diskusi tohoto tvrzení viz Pozza a spol. [206]).
Tyto výsledky tedy jednoznačně potvrzují, že takový stupeň hypoxie, jehož přítomnost v plicích in vivo je možno prežít [198], vyvolává v plicním endotelu in vitro přechodný vzrůst [Ca2+]i (což je známý intracelulární signál pro syntézu NO [76, 77, 78, 49, 79]) a zvýšené uvolňování NO do superfuzátu. Zdrojem tohoto kalciového signálu není influx extracelulárního Ca2+ přes plasmalemmu, nýbrž uvolnění Ca2+ ze sarkoplasmatického retikula. Důležitý je fakt, že zvýšená produkce NO plicním endotelem při akutní hypoxii byla změřena v systému, v němž nebyly přítomny hemodynamické důsledky hypoxie (změny tlaku, rychlosti proudu a střižného napětí). Hypoxie per se je tedy dostatečným stimulem pro aktivaci NO syntázy v plicním endotelu. To samozřejmě nevylučuje pravděpodobnost, že zvýšené sřrižné napětí při HPV in vivo dále potencuje syntézu NO [100, 146, 199, 102, 200, 201, 202]. Naopak, taková možnost je podporována skutečností, že inhibitory NO syntázy potencují nejen HPV, ale i odpověď na jiné vazokonstrikční podněty (např. angiotensin II) [89, 110, 91, 92, 119].
Jaký může být funkční význam zvýšené syntézy NO v plicním endotelu při akutní hypoxii? Nejpravděpodobnější je, že jde o mechanismus předcházející přílišnému zvýšení intravaskulárního tlaku, který by mohl poškodit tenkostěnné plicní cévy anebo zvýšit filtraci tekutiny z plicních cév do intersticia či do alveolů. Výsledný plicní edém by vážně ohrozil výměnu plynů. Podobná úloha je předpokládána pro prostacyklin, jehož uvolňování rovněž roste při akutní plicní vazokonstrikci, např. hypoxické [207]. Takovou úlohu NO podporuje výše zmíněný fakt, že inhibitory NO syntázy potencují odpověď na celou řadu vazokonstrikčních stimulů [89, 110, 91, 92, 119]. Navíc podle našich zkušeností je podstatně obtížnější uchránit izolované perfundované plíce vzniku alveolárního edému během vazokonstrikce, obsahuje-li perfuzát inhibitor NO syntázy.
Význam zvýšené syntézy NO oponující příliš silné plicní vazokonstrikci (kap. 5.3) výrazně ilustruje případ dexfenfluraminu.
Dexfenfluramin (distribuovaný u nás jako Isolipan, v USA jako Redux) byl vyvinut jako anorektikum. V průběhu posledního desetiletí se množily údaje o tom, že dexfenfluramin (nebo jeho racemická směs fenfluramin) může působit těžkou plicní hypertenzi [208, 209, 210, 211, 212, 213, 214]. V tom dexfenfluramin připomíná jiné anorektikum, aminorex, který na přelomu šedesátých a sedmdesátých let způsobil epidemii plicní hypertenze v západní Evropě [215, 216]. Nedávno byla publikována rozsáhlá studie z několika evropských center jednoznačně prokazující, že dexfenfluramin mnohonásobně zvyšuje pravděpodobnost onemocnění těžkou plicní hypertenzi [26]. Přitom se přes značné úsilí nedaří podáváním anorektik vyvolat výraznou chronickou plicní hypertenzi u laboratorních zvířat [217, 215, 218, 219]. To zřejmě souvisí s tím, že přestože dexfenfluramin zvyšuje pravděpodobnost onemocnění plicní hypertenzí, naprostá většina pacientů léčených dexfenfluraminem plicní hypertenzí neonemocní. Co rozhoduje o tom, kteří pacienti při užívání dexfenfluraminu plicní hypertenzi dostanou a kteří nikoliv, patří k nejdůležitejším nejasnostem týkajícím se plicní hypertenze vyvolané anorektiky.
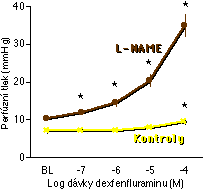
Naše zjištění, že plicní endotel brání syntézou NO příliš silné (zejména hypoxické) plicní vazokonstrikci (kap. 5.3), nás vedlo k úvaze, že plicní vazokonstrikční působení dexfenfluraminu může být ve zdravých plicích tlumeno syntézou NO. To by mohlo znamenat, že plicní hypertenzí onemocní při užívání dexfenfluraminu pouze ti pacienti, u nichž je plicní syntéza NO nějakým způsobem narušena. Tuto hypotézu jsme testovali měřením účinku L-NAME (5x10-5 M) na vazokonstrikční odpověď na dexfenfluramin v izolovaných perfundovaných plicích potkana [220]. Zjistili jsme, že v normálních, zdravých plicích má akutní podání dexfenfluraminu pouze velmi slabý vazokonstrikční účinek, a to pouze ve velmi vysoké dávce. Naproti tomu za přítomnosti L-NAME měl dexfenfluramin silný vazokonstrikční účinek již od poměrně nízkých dávek (Obrázek 5-4).
Toto zjištění, že stejně jako HPV je i plicní vazokonstrikční reaktivita na dexfenfluramin potencována inhibicí NO syntázy, je zvláště pozoruhodné s přihlédnutím na náš další nález, že totiž dexfenfluramin a hypoxie sdílí podstatnou část mechanismu, jímž působí vazokonstrikci.
Mechanismus HPV není dosud zcela jasný. Od roku 1992 je však zřejmé, že se v něm uplatňuje inhibice draslíkových (K) kanálů v membráně plicního cévního hladkého svalu [221]. Zajímalo nás, o jaké K kanály konkrétně jde, jakým mechanismem jsou v hypoxii inhibovány a jak jejich inhibice souvisí s vazokonstrikcí. Použili jsme metod terčíkového zamku v myocytech čerstvě enzymaticky izolovaných z proximálních a distálních plicních artérií/arteriol [222, 223]. Různé typy K kanálů byly rozlišeny pomocí selektivních inhibitorů. 4-aminopyridin (4-AP) byl použit jako poměrně selektivní inhibitor rodiny napětím řízených draslíkových (KV) kanálů (často nazývaných delayed rectifier) 10 [224]. Tetraetylamonium (TEA) a charybdotoxin inhibují přednostně vápníkem řízené draslíkové (KCa) kanály (charybdotoxin je v tomto smyslu podstatně selektivnější) [224].
Zjistili jsme [223], že v myocytech plicních cév hypoxie snižuje pravděpodobnost otevření K kanálu citlivého na 4-AP. V důsledku toho hypoxie redukuje celkový draslíkem nesený, 4-aminopyridem inhibovatelný elektrický proud napříč sarkolemmou (Obrázek 5-5). Biofyzikální charakterizace ukázala, že hypoxií inhibovaný kanál má poměrně malou vodivost 37 pS, což je - stejně jako citlivost na 4-AP - konzistentní s příslušnosti k rodině KV kanálů [224]. Naproti tomu KCa kanály (citlivé na TEA a charybdotoxin) s poměrně velkou vodivostí (170 pS) nebyly hypoxií ovlivněny.
Zcela novým druhem zjištění byla neuniformní distribuce různých typů K kanálu v plicním cévním řečišti. Kanály citlivé na hypoxii a 4-AP vysoce převládaly v myocytech izolovaných z distálních arteriol, zatímco KCa kanály převládaly v tepnách proximálních. To pozoruhodně koreluje se známou skutečností, že hlavním místem HPV jsou periferní plicní arterioly, zatímco velké plicní tepny se HPV neúčastní [17, 18, 19, 20, 21]. Význam neuniformní distribuce K kanálů pro umístění HPV jsme ostatně ověřili na izolovaných cévních kroužcích in vitro. Kroužky izolované z plícnice odpovídaly na hypoxii slabou konstrikcí, kterou do 5 minut nahradila výrazná vazodilatace. Naproti tomu kroužky izolované z distálních plicních arteriol odpovídaly na hypoxii výraznější a hlavně setrvalou konstrikcí, kterou neovlivnilo TEA, avšak značně ji potencoval 4-AP.
Zjistili jsme také, že K kanály citlivé na 4-AP jsou rozhodující pro řízení klidového membránového potenciálu myocytů periferních plicních arteriol. 4-AP totiž působil výraznou depolarizaci, zatímco TEA i charybdotoxin byly bez účinku. Podobnou depolarizaci jako 4-AP působila hypoxie.
Všechny tyto údaje dohromady potvrzují hypotézu, podle níž hypoxie působí periferní plicní vazokonstrikci tím, že inhibuje KV kanál zodpovědný za udržování membránového potenciálu. To vede k depolarizaci a následné aktivaci napětím řízených vápníkových kanálů [96], influxu vápníku [225] a aktivaci kontraktilního aparátu zvýšenou [Ca2+]i [226].
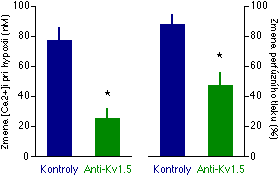
Ve snaze dále definovat, který přesně kanál ze skupiny KV kanálů je zodpovědný za HPV, jsme testovali protilátky proti jednomu z vážných kandidátů, kanálu zvanému Kv1.5 [227]. Měřili jsme vliv komerčně vyráběných polyklonálních anti-Kv1.5 protilátek (Upstate Biotechnology Inc.) na vzrůst [Ca2+]i při hypoxii v myocytech izolovaných z odporových plicních arteriol potkana a na HPV v izolovaných perfundovaných plicích potkana [228, 229]. Zjistili jsme, že anti-Kv1.5 protilátky inhibovaly odpověď na hypoxii v obou těchto systémech (Obrázek 5-6), zatímco protilátky proti jinému KV kanálu (Kv2.1) nikoliv [228, 229]. Reaktivita na jiné vazokonstrikční podněty (angiotensin II) nebyla anti-Kv1.5 protilátkami ovlivněna. Tyto nálezy jsou konzistentní s hypotézou, že K kanálem, citlivým na hypoxii v plicních cévách, je Kv1.5.
Mechanismus, jakým působí plicní hypertenzi dexfenfluramin, byl zcela neznámý. Naše pokusy ukazují, že dexfenfluramin inhibuje draslíkem nesený proud sarkolemmou myocytů izolovaných z periferních plicních arteriol [220]. Stejně jako v případě hypoxie je tento proud inhibovatelný podáním 4-AP (Obrázek 5-5), avšak nikoliv TEA. Podobně jako hypoxie působí dexfenfluramin mebránovou depolarizaci, influx vápníku a vzrůst [Ca2+]i [230]. Je tedy velmi pravděpodobné, že hypoxie a dexfenfluramin sdílí podstatnou část mechanismu, jímž působí plicní vazokonstrikci. V obou případech se endogenní NO vazokonstrikčního mechanismu přímo neúčastní, avšak významným způsobem výslednou odpověď moduluje (Obrázek 5-1 a Obrázek 5-4).
Výsledky popsané v kapitole 5 (shrnuje je Tabulka 5-2) dohromady ukazují, že při HPV roste uvolňování NO z plicního endotelu. Stimulem je jak vazokonstrikce, tak hypoxie per se. Naše nálezy o úloze NO při HPV jsou podkladem našeho příspěvku do monografie o úloze NO v plicích [65].
Tabulka 5-2: Souhrn výsledků ukazujících zvýšenou plicní tvorbu NO při hypoxii a/nebo vazokonstrikci
| 1. | Inhibitory NO syntázy potencuji plicní vazokonstrikci (i hypoxickou). | |
| 2. | Hladina NO a jeho oxidačního produktu NO2- v superfuzátu kultury plicních arteriálních endoteliálních buněk roste při akutní hypoxii |
[125]
|
| 3. | Akutní hypoxie vyvolává v plicních endoteliálních buňkách signál pro tvorbu NO (zvýšení [Ca2+]i) |
[125]
|
Jak už bylo zmíněno (kap. 5.1), nás nález, že inhibitory NO syntázy potencují plicní vazokonstrikci včetně HPV (Obrázek 5-1), je v souladu s mnoha jinými laboratořemi [149, 90, 110, 91, 193, 194, 195, 113, 196, 114, 117, 139, 118, 146, 147, 151, 197]. Naše měření akumulace NO+ NO2- v superfuzátu endoteliálních buněk při hypoxii (Obrázek 5-2) je naproti tomu unikátní. Shaul a Wellsová [231] provedli podobný pokus, neměřili však NO ani NO2-, nýbrž cGMP v hladkém svalu kultivovaném společně s fetálním plicním endotelem. cGMP zprostředkuje vazodilatační působení NO, jeho intracelulární hladina je však kromě NO ovlivňována ještě celou řadou dalších stimulů. Při snížení PO2 ze 150 na 40 mmHg byla patrná mírná tendence ke vzrůstu bazální hladiny cGMP, která však nedosáhla statistické významnosti [231]. V podobném pokuse popsala stejná skupina snížení cGMP při akutní hypoxii [232]; zde však byly použity izolované segmenty plícnice, která je z hlediska zvýšení plicního hemodynamického odporu při působení akutní hypoxie nevýznamná [17, 18, 19, 20, 21].
Grimminger a spolupracovníci [118] měřili akumulaci NO+ NO2- v perfuzátu izolovaných plic králíka při akutní hypoxii a nenalezli žádnou změnu. Ve vydechovaném vzduchu ubylo při hypoxii NO, avšak podle jejich i našeho názoru nešlo o NO pocházející z plicních cév, nýbrž z dýchacích cest. Elegantním potvrzením našeho tvrzení o tlumení HPV in vivo endoteliální syntézou NO je nedávno publikovaný pokus Janssense a spolupracovníků [233]. Ukazali, že markantní zvýšení obsahu endoteliální NO syntázy v myších plicích, navozené adenovirovým transferem genu pro lidskou endoteliální NO syntázu, značně redukuje HPV.
| Předchozí kapitola |
|
17. Kato M, Staub NC: Response of small pulmonary arteries to unilobar hypoxia and hypercapnia. Circ Res 1966; 19: 426-439.
18. Nagasaka K, Bhattacharya J, Nanjo S, Gropper MA, Staub NC: Micropuncture measurement of lung microvascular pressure profile during hypoxia in cats. Circ Res 1984; 54: 90-95.
19. Madden JA, Dawson CA, Harder DR: Hypoxia-induced activation in small isolated pulmonary arteries from the cat. J Appl Physiol 1985; 59: 113-118.
20. Shirai M, Sada K, Ninomiya I: Effects of regional alveolar hypoxia and hypercapnia on small pulmonary vessels in cats. J Appl Physiol 1986; 61: 440-448.
21. Madden JA, Vadula MS, Kurup VP: Effects of hypoxia and other vasoactive agents on pulmonary and cerebral artery smooth muscle cells. Am J Physiol 1992; 263: L384-L393.
26. Abenhaim L, Moride Y, Brenot F, Rich S, Benichou J, Kurz X, Higenbottam T, Oakley C, Wouters E: Appetite-suppressant drugs and the risk of primary pulmonary hypertension. N Engl J Med 1996; 335: 609-616.
47. Palmer MJ, Moncada S: A novel citrulline-forming enzyme implicated in the formation of nitric oxide by vascular endothelial cells. Biochem Biophys Res Commun 1989; 158: 348-352.
48. Leone AM, Palmer RM, Knowles RG, Francis PL, Ashton DS, Moncada S: Constitutive and inducible nitric oxide synthases incorporate molecular oxygen into both nitric oxide and citrulline. J Biol Chem 1991; 266: 23790-23795.
49. Nathan C: Nitric oxide as a secretory product of mammalian cells. FASEB J 1992; 6: 3051-3064.
61. Archer SL, Hampl V, Huang J, Cowan N: The importance of calcium in the regulation of EDRF synthesis in the pulmonary vasculature. In: Weir EK, Hume JR, Reeves JT, Eds. Ion Flux in Pulmonary Vascular Control. New York, Plenum, 1993; 223-246.
65. Hampl V, Archer SL: The role of endogenous nitric oxide in acute hypoxic pulmonary vasoconstriction. In: Zapol WM, Bloch KD, Eds. Nitric Oxide and the Lung. New York, Marcel Dekker, 1997; 113-135.
76. Johns A, Lategan TW, Lodge NJ, Ryan US, van Breemen C, Adams DJ: Calcium entry through receptor operated channels in bovine pulmonary artery endothelial cells. Tissue and Cell 1987; 19: 733-745.
77. Peach MJ, Singer HA, Izzo NJ, Loeb AL: Role of calcium in endothelium-dependent relaxation of arterial smooth muscle. Am J Cardiol 1987; 59: A35-A43.
78. Luckhoff A, Pohl U, Mulsch A, Busse R: Differential role of extra- and intracellular calcium in the release of EDRF and prostacyclin from cultured endothelial cells. Br J Pharmacol 1988; 95: 189-196.
79. Blatter LA, Taha Z, Mesaros S, Shacklock PS, Wier WG, Malinski T: Simultaneous measurements of Ca2+ and nitric oxide in bradykinin-stimulated vascular endothelial cells. Circ Res 1995; 76: 922-924.
89. Archer SL, Tolins JP, Raij L, Weir EK: Hypoxic pulmonary vasoconstriction is enhanced by inhibition of the synthesis of an endothelium derived relaxing factor. Biochem Biophys Res Commun 1989; 164: 1198-1205.
90. Robertson BE, Warren JB, Nye PCG: Inhibition of nitric oxide synthesis potentiates hypoxic vasoconstriction in isolated rat lungs. Exp Physiol 1990; 75: 255-257.
91. Liu S, Crawley DE, Barnes PJ, Evans TW: Endothelium-derived relaxing factor inhibits hypoxic pulmonary vasoconstriction in rats. Am Rev Respir Dis 1991; 143: 32-37.
92. Hampl V, Archer SL, Nelson DP, Weir EK: Chronic EDRF inhibition and hypoxia: effects on pulmonary circulation and systemic blood pressure. J Appl Physiol 1993; 75: 1748-1757. (abstrakt)
96. McMurtry IF, Davidson AB, Reeves JT, Grover RF: Inhibition of hypoxic pulmonary vasoconstriction by calcium antagonists in isolated rat lungs. Circ Res 1976; 38: 99-104.
100. Hecker M, Mulsch A, Bassenge E, Busse R: Vasoconstriction and increased flow: two principal mechanisms of shear stress-dependent endothelial autacoid release. Am J Physiol 1993; 265: H828-H833.
102. Kuchan MJ, Jo H, Frangos JA: Role of G proteins in shear stress-mediated nitric oxide production by endothelial cells. Am J Physiol 1994; 267: C753-C758.
110. Hasunuma K, Yamaguchi T, Rodman DM, O'Brien RF, McMurtry IF: Effects of inhibitors of EDRF and EDHF on vasoreactivity of perfused rat lungs. Am J Physiol 1991; 260: L97-L104.
113. Barer G, Emery C, Stewart A, Bee D, Howard P: Endothelial control of the pulmonary circulation in normal and chronically hypoxic rats. J Physiol 1993; 463: 1-16.
114. Oka M, Hasunuma K, Webb SA, Stelzner TJ, Rodman DM, McMurtry IF: EDRF suppresses an unidentified vasoconstrictor mechanism in hypertensive rat lungs. Am J Physiol 1993; 264: L587-L597.
117. Zhao L, Crawley DE, Hughes JMB, Evans TW, Winter RJD: Endothelium-derived relaxing factor activity in rat lung during hypoxic pulmonary vascular remodeling. J Appl Physiol 1993; 74: 1061-1065.
118. Grimminger F, Spiestersbach R, Weismann N, Seeger W: Nitric oxide generation and hypoxic vasoconstriction in buffer-perfused rabbit lungs. Am. J. Respir. Crit. Care Med. 1994; 149: A21.
119. Isaacson TC, Hampl V, Weir EK, Nelson DP, Archer SL: Increased endothelium-derived nitric oxide in hypertensive pulmonary circulation of chronically hypoxic rats. J Appl Physiol 1994; 76: 933-940. (abstrakt)
125. Hampl V, Cornfield DN, Cowan NJ, Archer SL: Hypoxia potentiates nitric oxide synthesis and transiently increases cytosolic calcium levels in pulmonary artery endothelial cells. Eur Respir J 1995; 8: 515-522. (abstrakt)
126. Hampl V, Cowan NJ, Archer SL: Acute hypoxia increases nitric oxide synthesis in cultured pulmonary arterial cells. Am. J. Respir. Crit. Care Med. 1995; 151: A627.
139. Dumas JP, Dumas M, Sgro C, Advenier C, Giudicelli JF: Effects of two K+ channel openers, aprikalim and pinacidil, on hypoxic pulmonary vasoconstriction. Eur J Pharmacol 1994; 263: 17-23.
146. Hakim TS: Flow-induced release of EDRF in the pulmonary vasculature: site of release and action. Am J Physiol 1994; 267: H363-H369.
147. Leeman M, De Beyl VZ, Delcroix M, Naeije R: Effects of endogenous nitric oxide on pulmonary vascular tone in intact dogs. Am J Physiol 1994; 266: H2343-H2347.
149. Persson MG, Gustafsson LE, Wiklund NP, Moncada S, Hedqvist P: Endogenous nitric oxide as a probable modulator of pulmonary circulation and hypoxic pressor response in vivo. Acta Physiol Scand 1990; 140: 449-457.
151. Persson MG, Kalzen H, Gustafsson LE: Oxygen or low concentrations of nitric oxide reverse pulmonary vasoconstriction induced by nitric oxide synthesis inhibition in rabbits. Acta Physiol Scand 1994; 150: 405-411.
157. van Gelderen EM, Den Boer MO, Saxena PR: NG-nitro l-arginine methyl ester: systemic and pulmonary haemodynamics, tissue blood flow and arteriovenous shunting in the pig. Naunyn-Schmiedeberg's Arch Pharmacol 1993; 348: 417-423.
158. McMahon TJ, Hood JS, Bellan JA, Kadowitz PJ: Nomega-nitro-L-arginine methyl ester selectively inhibits pulmonary vasodilator responses to acetylcholine and bradykinin. J Appl Physiol 1991; 71: 2026-2031.
159. Stamler JS, Loh E, Roddy M-A, Currie KE, Creager MA: Nitric oxide regulates basal systemic and pulmonary vascular resistance in healthy humans. Circulation 1994; 89: 2035-2040.
160. Blitzer ML, Loh E, Roddy MA, Stamler JS, Creager MA: Endothelium-derived nitric oxide regulates systemic and pulmonary vascular resistance during acute hypoxia in humans. J Am Coll Cardiol 1996; 28: 591-596.
187. Rengasamy A, Johns RA: Characterization of endothelium-derived relaxing factor/nitric oxide synthase from bovine cerebellum and mechanism of modulation by high and low oxygen tensions. J Pharmacol Exp Ther 1991; 259: 310-316.
188. Johns RA, Linden JM, Peach MJ: Endothelium-dependent relaxation and cyclic GMP accumulation in rabbit pulmonary artery are selectively impaired by moderate hypoxia. Circ Res 1989; 65: 1508-1515.
189. Warren JB, Maltby NH, McCormack D, Barnes PJ: Pulmonary endothelium-derived relaxing factor is impaired in hypoxia. Clin Sci 1989; 77: 671-676.
190. Rodman DM, Yamaguchi T, Hasunuma K, O'Brien R, McMurtry IF: Effects of hypoxia on endothelium-dependent relaxation of rat pulmonary artery. Am J Physiol 1990; 258: L207-L214.
191. Demiryurek AT, Wadsworth RM, Kane KA, Peacock AJ: The role of endothelium in hypoxic constriction of human pulmonary artery rings. Am Rev Respir Dis 1993; 147: 283-290.
192. Isaacson T, Weir EK, Hampl V, Nelson D, Archer S: Enhanced vasoconstriction in chronic hypoxic pulmonary hypertension is not associated with reduced responsiveness to EDRF or nitric oxide. FASEB J. 1992; 6: A1177.
193. Fineman JR, Chang R, Soifer SJ: EDRF inhibition augments pulmonary hypertension in intact newborn lambs. Am J Physiol 1992; 262: H1365-H1371.
194. Ogata M, Ohe M, Katayose D, Takishima T: Modulatory role of EDRF in hypoxic contraction of isolated porcine pulmonary arteries. Am J Physiol 1992; 262: H691-H697.
195. Sprague RS, Thiemermann C, Vane JR: Endogenous endothelium-derived relaxing factor opposes hypoxic pulmonary vasoconstriction and supports blood flow to hypoxic alveoli in anesthetized rabbits. Proc Natl Acad Sci USA 1992; 89: 8711-8715.
196. McCormack DG, Paterson NAM: Loss of hypoxic pulmonary vasoconstriction in chronic pneumonia is not mediated by nitric oxide. Am J Physiol 1993; 265: H1523-H1528.
197. Fredťn F, Wei SZ, Berglund JE, Frostell C, Hedenstierna G: Nitric oxide modulation of pulmonary blood flow distribution in lobar hypoxia. Anesthesiology 1995; 82: 1216-1225.
198. Groves BM, Reeves JT, Sutton JR, Wagner PD, Cymerman A, Malconian MK, Rock PB, Young PM, Houston CS: Operation Everest II: elevated high-altitude pulmonary resistance unresponsive to oxygen. J Appl Physiol 1987; 63: 521-530.
199. Korenaga R, Ando J, Tsuboi H, Yang W, Sakuma I, Toyo-oka T, Kamiya A: Laminar flow stimulates ATP- and shear stress-dependent nitric oxide production in cultured bovine endothelial cells. Biochem Biophys Res Commun 1994; 198: 213-219.
200. Kanai AJ, Strauss HC, Truskey GA, Crews AL, Grunfeld S, Malinski T: Shear stress induces ATP-independent transient nitric oxide release from vascular endothelial cells, measured directly with a porphyrinic microsensor. Circ Res 1995; 77: 284-293.
201. Melkumyants AM, Balashov SA, Khayutin VM: Control of arterial lumen by shear stress on endothelium. News Physiol Sci 1995; 10: 204-210.
202. Corson MA, James NL, Latta SE, Nerem RM, Berk BC, Harrison DG: Phosphorylation of endothelial nitric oxide synthase in response to fluid shear stress. Circ Res 1996; 79: 984-991.
203. Grynkiewicz G, Poenie M, Tsien RY: A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 1985; 260: 3440-3450.
204. Meissner G, el-Hashem A: Ryanodine as a functional probe of the skeletal muscle sarcoplasmic reticulum Ca2+ release channel. Mol Cell Biochem 1992; 114: 119-123.
205. Inesi G, Sagara Y: Thapsigargin, a high affinity and global inhibitor of intracellular Ca2+ transport ATPases. Arch Biochem Biophys 1992; 298: 313-317.
206. Pozzan T, Rizzuto R, Volpe P, Meldolesi J: Molecular and cellular physiology of intracellular calcium stores. Physiol Rev 1994; 74: 595-636.
207. Voelkel NF, Gerber JG, McMurtry IF, Nies AS, Reeves JT: Release of vasodilator prostaglandin, PGI2, from isolated rat lung during vasoconstriction. Circ Res 1981; 48: 207-213.
208. Fahlen M, Bergman H, Helder G, Ryden L, Wallentin I, Zettergren L: Phenformin and pulmonary hypertension. Br Heart J 1973; 35: 824-828.
209. Douglas J, Munro J, Kitchin A, Muir A, Proudfoot A: Pulmonary hypertension and fenfluramine. Brit Med J 1981; 283: 881-883.
210. Gaul G, Blazek G, Deutsch E, Heeger H: Ein Fall von chronischer pulmonar Hypertonie nach Fenfluramine inname. Wiener Klinische Wochenschrift 1982; 94: 618-622.
211. Loogen F, Worth H, Schwan G, Goeckenjan G, Losse B, Horstkotte D: Long-term follow-up of pulmonary hypertension in patients with and without anorectic drug intake. Cor et Vasa 1985; 27: 111-124.
212. Atanassoff PG, Weiss BM, Schmid ER, Tornic M: Pulmonary hypertension and dexfenfluramine. Lancet 1992; 339: 436.
213. Roche N, Labrune S, Braun JM, Huchon GJ: Pulmonary hypertension and dexfenfluramine. Lancet 1992; 339: 436-437.
214. Brenot F, Herve P, Petitpretz P, Parent F, Duroux P, Simonneau G: Primary pulmonary hypertension and fenfluramine use. Br Heart J 1993; 70: 537-541.
215. Kay JM, Smith P, Heath D: Aminorex and the pulmonary circulation. Thorax 1971; 26: 262-270.
216. Gurtner HP: Aminorex and pulmonary hypertension. Cor et Vasa 1985; 27: 160-171.
217. Engelhardt A, Kroneberg G, Stoepel K, Stotzer H: The effects of acute and chronic administration of sympathomimetic substances on the systemic and pulmonary circulation. Proc Eur Soc Drug Toxicol 1970; 11: 110-117.
218. Byrne-Wuinn E, Grover R: Aminorex (Menocil) and amphetamine: acute and chronic effects on pulmonary and systemic haemodynamics in the calf. Thorax 1972; 27: 127-131.
219. Smith P, Heath D, Kay J, Wright J, McKendrick C: Pulmonary arterial pressure and structure in the Pats monkey after prolonged administration of aminorex fumarate. Cardiovasc Res 1973; 7: 30-38.
220. Weir EK, Reeve HL, Huang JMC, Michelakis E, Nelson DP, Hampl V, Archer SL: Anorexic agents aminorex, fenfluramine, and dexfenfluramine inhibit potassium current in rat pulmonary vascular smooth muscle and cause pulmonary vasoconstriction. Circulation 1996; 94: 2216-2220. (abstrakt)
221. Post JM, Hume JR, Archer SL, Weir EK: Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. Am J Physiol 1992; 262: C882-C890.
222. Archer SL, Reeve HL, Hampl V, Huang J, Peterson DA, Weir EK: Redox regulation of pulmonary vascular tone: a central role for potassium channels. J. Physiol. 1995; 487.P: 11S-12S.
223. Archer SL, Huang JMC, Reeve HL, Hampl V, Tolarová S, Michelakis E, Weir EK: Differential distribution of electrophysiologically distinct myocytes in conduit and resistance arteries determines their response to nitric oxide and hypoxia. Circ Res 1996; 78: 431-442. (abstrakt)
224. Nelson MT, Quayle JM: Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol 1995; 268: C799-C822.
225. Vadula MS, Kleinman JG, Madden JA: Effect of hypoxia and norepinephrine on cytoplasmic free Ca2+ in pulmonary and cerebral arterial myocytes. Am J Physiol 1993; 265: L591-L597.
226. McCall D: Excitation-contraction coupling in cardiac and vascular smooth muscle: modification by calcium-entry blockade. Circulation 1987; 75: 3-14.
227. Takimoto K, Levitan ES: Glucocorticoid induction of Kv1.5 K+ channel gene expression in ventricle of rat heart. Circ Res 1994; 75: 1006-1013.
228. Archer SL, Souil E, Dinh-Xuan AT, Schremmer B, Mercier J-C, El Yaagoubi A, Nguyen-Huu L, Reeve HL, Hampl V: Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and 2.1 in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J Clin Invest 1998; 101: 2319-2330.
229. Hampl V, Nelson DP, Archer SL: Antibodies against the Kv1.5 voltage-gated potassium channel inhibit hypoxic pulmonary vasoconstriction. Physiol. Res. 1997; 65: P28.
230. Reeve HL, Archer SL, Soper M, Weir EK: Dexfenfluramine increases pulmonary artery smooth muscle intracellular Ca2+, independent of membrane potential. Am J Physiol 1999; 277: L662-L666.
231. Shaul PW, Wells LB: Oxygen modulates nitric oxide production selectively in fetal pulmonary endothelial cells. Am J Respir Cell Mol Biol 1994; 11: 432-438.
232. Shaul PW, Wells LB, Horning KM: Acute and prolonged hypoxia attenuate endothelial nitric oxide production in rat pulmonary arteries by different mechanisms. J Cardiovasc Pharmacol 1993; 22: 819-827.
233. Janssens SP, Bloch KD, Nong ZX, Gerard RD, Zoldhelyi P, Collen D: Adenoviral-mediated transfer of the human endothelial nitric oxide synthase gene reduces acute hypoxic pulmonary vasoconstriction in rats. J Clin Invest 1996; 98: 317-324.
10 Průchod elektrického proudu jakýmkoliv vodičem, tedy i iontovým kanálem, je úměrný napětí. Napětím řízeným kanálem však zde rozumíme takový, jehož kinetika (otevírání, zavírání, inaktivace) je řízena napětím.
| Předchozí kapitola |
|